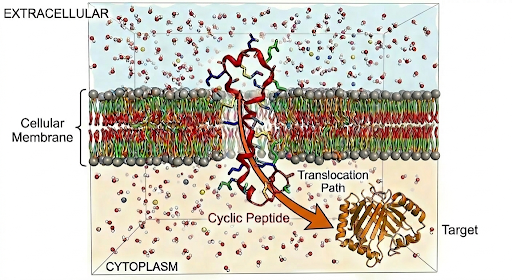
Building on the potential of cyclic peptides to target “undruggable” intracellular protein–protein interactions, the research focuses on overcoming a critical limitation: poor membrane permeability.
Traditional experimental methods to assess permeability are both time-consuming and resource-intensive, while existing computational approaches often lack sufficient predictive accuracy. This creates a bottleneck in identifying viable drug candidates.
By integrating large-scale MD simulations with machine learning models, researchers are able to better understand how cyclic peptides behave. This allows them to identify promising candidates earlier and prioritise them for experimental validation, improving the efficiency of the drug discovery process.
This work was enabled by NSCC Singapore’s ASPIRE 2A+ supercomputer, which provided the computational scale required for data generation, simulation and AI model development.
The research was enabled through close collaboration across industry, academia and high-performance computing (HPC). WuXi AppTec, a Contract Research, Development and Manufacturing Organisation (CRDMO), synthesised the cyclic peptides and measured membrane permeability using the Parallel Artificial Membrane Permeability Assay (PAMPA), providing experimental data to validate computational predictions. Chosun University (Republic of Korea) contributed specialised expertise in computational physics, supporting the development of the LUFnet model.
At the core of this project is the integration of high performance computing with artificial intelligence (AI) to model molecular behaviour at scale. Using NSCC Singapore’s ASPIRE 2A+ supercomputer, researchers modelled the dynamic behaviours of over 5,000 cyclic peptides across different environments.
Simulations were conducted in both water and hexane to capture how these molecules adapt their structure across environments, allowing researchers to evaluate their likelihood of crossing cell membranes.
To efficiently process the large volume of data, the team developed LUFnet, a scalable AI model designed to enhance molecular dynamics (MD) simulations. By enabling larger time integration steps while preserving key physical constraints, LUFnet allows l molecular motion to be simulated over longer timescales without compromising accuracy.
Through the close integration of HPC and AI, the project delivered several major innovations: the LUFnet architecture, a first-of-its-kind dataset called CycPeptMPDB-4D, and the first rigorous benchmark evaluating 13 different AI models for permeability prediction.
NSCC Singapore’s ASPIRE 2A+ supercomputer was critical in enabling this research, providing the scale and the performance required to integrate large-scale simulations with AI model development.
- Large-scale MD simulations: Over 10,000 independent GROMACS simulations were executed on ASPIRE 2A+ to generate the CycPeptMPDB-4D dataset
- Deep learning model training: ASPIRE 2A+’s GPU nodes enabled the intensive training and benchmarking of 13 AI models across diverse data splits and hyperparameter configurations
- Accelerated simulation development: Parallel computing capabilities of ASPIRE 2A+ made it feasible to generate high-fidelity reference trajectories and train the LUFnet transformer across multiple thermodynamic states and system sizes. Simulations were run 5 times faster than other conventional research methods, reducing half-year workloads to just one month.
- Large-scale MD simulations: Over 10,000 independent GROMACS simulations were executed on ASPIRE 2A+ to generate the CycPeptMPDB-4D dataset
- Streamlined research workflow: Simulation, data generation and AI model training were conducted within a unified environment, eliminating the need for cross-system data transfer.
These capabilities allowed researchers to explore thousands of molecular candidates, capture complex molecular behaviour with greater fidelity, and conduct analyses that would not have been feasible using conventional computing resources.
The research introduces a more targeted and computationally driven approach to discovering cell-permeable cyclic peptides — addressing a key challenge in developing therapies for diseases such as cancer that involve intracellular protein–protein interactions.
By combining AI models, scalable simulation methods and large-scale datasets, the project establishes an integrated framework that can be applied beyond cyclic peptides. These approaches can be applied to predict other critical molecular properties, such as oral bioavailability and metabolic stability — key factors in the successful development of new therapeutics.
The outputs of this research, including the CycPeptMPDB-4D dataset, LUFnet simulation framework, and AI benchmarking approaches are openly available. This supports broader adoption across the global research and pharmaceutical community and enables more efficient, data-driven drug discovery. Based on this dataset, the team is also developing their own AI model.
Beyond the research community, these advances contribute to ongoing efforts to develop new treatment strategies for diseases that are currently difficult to target, while improving the efficiency of early-stage drug discovery.
Looking ahead, the availability of these datasets, benchmarks and tools is expected to support near-term adoption across the research and pharmaceutical landscape. Over time, this foundation can be extended through generative AI models capable of designing cyclic peptides with optimised permeability and target specificity.
In the longer term, the project contributes towards a more integrated computational pipeline for cyclic peptide drug discovery — from initial target identification through to the selection of viable clinical candidates.
Supported by a strong collaborative network across industry and academia, the team is extending these AI-driven tools into new therapeutic areas, establishing a more resource-efficient pathway for drug development.
“Cyclic peptides change conformation as they cross membranes, so capturing that structural flexibility is key to predicting permeability. This involves modelling thousands of molecular dynamics trajectories and training AI models on large-scale datasets — tasks that go far beyond the capabilities of desktop computing. NSCC Singapore’s ASPIRE 2A+ supercomputing resources were instrumental in enabling this work, providing the computational power needed to perform these simulations efficiently and at scale.”

Liu Wei
Senior Scientist II
A*STAR Bioinformatics Institute (BII)